The new EU In-vitro Diagnostic Regulation (Regulation EU 2017/546, IVDR) is a European Union regulation that governs the requirements for in-vitro diagnostic (IVD) devices. It entered into force on May 26, 2017, replacing the previous EU Directive on in-vitro diagnostic medical devices (Directive 98/79/EC, IVDD). Manufacturers had a transition period of five years, until May 26, 2022, to implement the revised IVDR requirements for CE-marked in-vitro diagnostic devices. The regulation is legally enforceable in all member states of the European Union and the European Free Trade Association (EFTA).
IVDR Requirements
The IVDR places more stringent requirements on manufacturers to ensure the safety, performance, and usefulness (known as performance evaluation) of devices. Among other things, the regulation requires rigorous review of technical documentation and clinical evidence to ensure that IVD products meet essential safety and performance requirements. Manufacturers must also demonstrate a quality management system and conduct ongoing risk assessments, as well as demonstrate the lifetime safety of their products.
The IVDR also brings changes for Notified Bodies, which are responsible for approving IVD products. Notified Bodies must now meet higher requirements and will be monitored by national authorities on a regular basis. The monitoring of IVD products will be more stringent as well. For example, from now on, a Notified Body must be involved for products in risk class B (medium risk level) and higher (risk classes C and D). This automatically increases the number of products that need to be monitored. Thus, the IVDR represents a major challenge for IVD product manufacturers, but also offers benefits for patients’ safety and the healthcare system. The stricter requirements for IVD products will help ensure that only high quality and safe products will be placed on the market. This, in turn, will help ensure that patients receive a more accurate diagnosis and that treatment can be adjusted accordingly.
IVDR Classification
All in-vitro diagnostic devices are classified into classes A, B, C or D according to IVDR, instead of two lists (A and B) so far. The classification process is mainly based on the intended purpose of the devices and the resulting expected risks.
It is now very important to know the class of the device at early stages of development, because under the new regulations the applicable conformity assessment procedure changes depending on the class.
You can find further information on the correct classification of medical devices and in-vitro diagnostic medical devices here.
Transition Plan
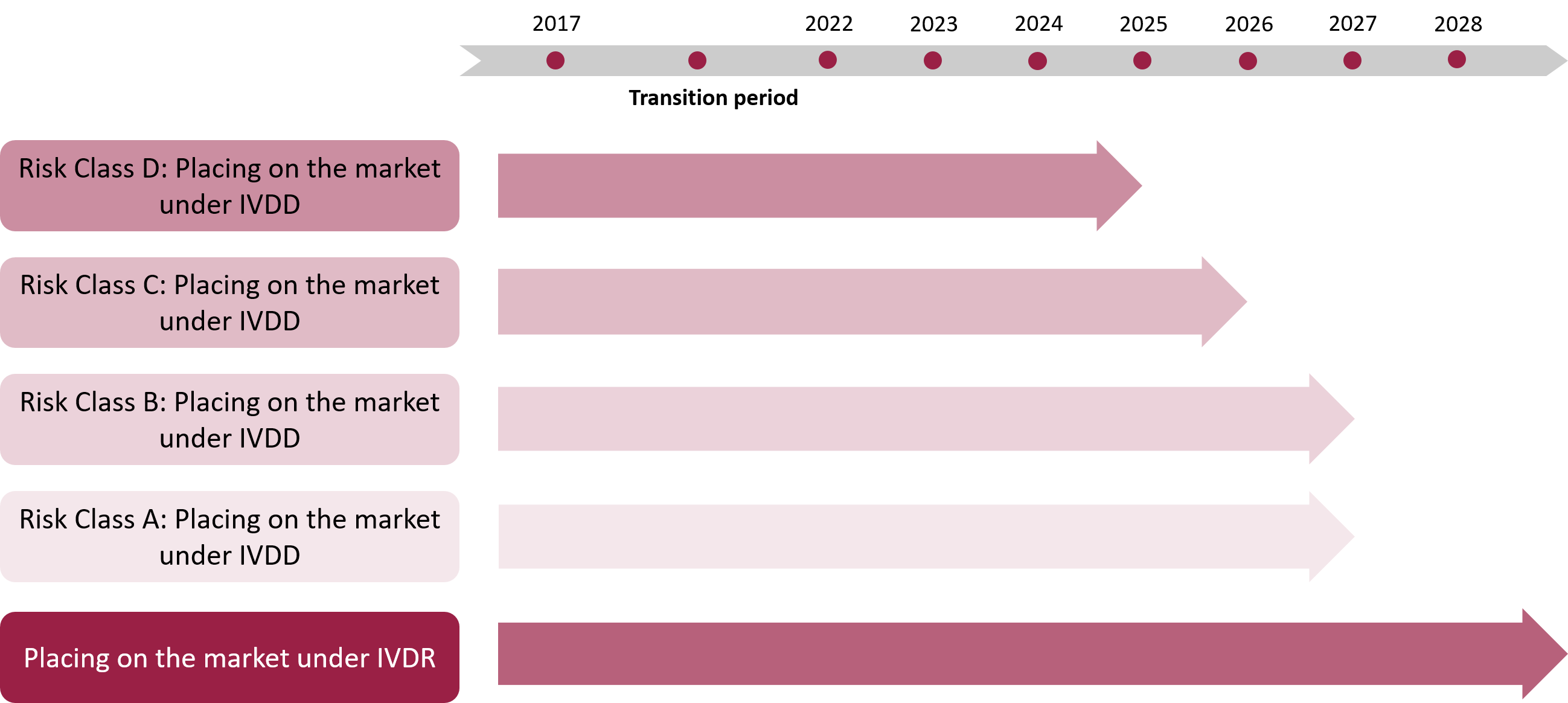
In accordance with Regulation (EU) 2023/607 of the European Parliament and of the Council of 15 March 2023, the transitional provisions for in-vitro diagnostic medical devices have been adjusted to prevent supply shortages and potential disruptions in healthcare systems.
The transitional periods outlined in Regulation (EU) 2023/607 allow products that complied with the requirements of the previous In-Vitro Diagnostic Directive (IVDD) to continue being placed on the market or put into service under certain conditions, while they are progressively adapted to the requirements of Regulation (EU) 2017/746 (IVDR).
The transitional periods depend on the classification of the products as defined in Regulation (EU) 2017/746 and are subject to specific conditions:
Products placed on the EU market before 26 May 2022:
- Products without the involvement of a Notified Body (self-declared):
- 31 December 2027: End of the transition period for Class D products.
- 31 December 2028: End of the transition period for Class C products.
- 31 December 2029: End of the transition period for Class B products and Class A sterile products.
- Products certified by a Notified Body:
- These may continue to be placed on the market or used until 31 December 2027, provided that the conditions for the extended transition period are met.
Conditions for using the extended transition period:
- By 26 May 2025: Implementation of a quality management system (QMS) compliant with the IVDR.
- By 26 May 2025, 2026, or 2027: Submission of an application for conformity assessment.
- By 26 September 2025, 2026, or 2027: Conclusion of a written agreement with a Notified Body and transfer of surveillance.
- No significant changes to the design or intended purpose.
- Continued compliance with the requirements of the previous EU legislation (IVDD).
- Compliance with the requirements of Directive 98/79/EC (IVDD).
Source:
European Union (2023). Regulation (EU) 2023/607 of the European Parliament and of the Council of 15 March 2023 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards the transitional provisions for certain medical devices and in-vitro diagnostic medical devices. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=OJ:L_202401860.