Die Europäische Verordnung für Medizinprodukte (Medical Device Regulation, EU MDR) ist am 25. Mai 2017 in Kraft getreten. Zusammen mit der EU-Verordnung über In-vitro-Diagnostika (In-vitro-Diagnostic Device Regulation, IVDR) von 2017 ersetzten die MDR am 26. Mai 2021 bzw. die IVDR am 26. Mai 2022 die bestehenden Richtlinien. Die MDR hat zum Ziel, ein hohes Niveau an Sicherheit und Gesundheitsschutz für Patienten, Anwender und Dritte zu gewährleisten sowie den freien Verkehr von Medizinprodukten im Binnenmarkt zu fördern.
Gemäß der sich ändernden Richtlinien werden künftig die Anforderungen für die Zertifizierung von Medizinprodukten im europäischen Gesundheitswesen anhand strengeren und einheitlichen Regeln erfolgen.
Dies hat Auswirkungen auf alle Hersteller von Medizinprodukten, welche erstmalig eine Zertifizierung innerhalb der EU anstreben oder deren Produkte sich bereits erfolgreich am Markt etabliert haben.
Auf unserer Webseite finden Hersteller und andere Akteure im Medizinproduktebereich Hilfe und umfangreiche Informationen zu den Veränderungen, welche die MDR mit sich bringt. Weitere Hintergrundinformationen zu der IVDR oder zu Digitalen Gesundheitsanwendungen (DiGA) finden Sie unter den entsprechenden Links.
Um Ihnen den Übergang zur MDR oder IVDR zu erleichtern, bietet Ihnen das Medical Device Innovation Center (MIC) in Mainz umfassende Unterstützung in allen Aspekten an – von der Klassifizierung Ihrer Produkte über die Anpassung technischer Dokumentationen bis hin zur Durchführung der Konformitätsbewertung. Wir arbeiten eng mit Ihnen und unseren Partnern zusammen, um eine individuelle Lösung für Ihre spezifischen Bedürfnisse zu finden.
Kontaktieren Sie uns noch heute unter info@mic-mainz.de oder +49 6131 17-9646 und lassen Sie sich umfassend von unseren Expert:innen beraten.
Was ist die MDR?
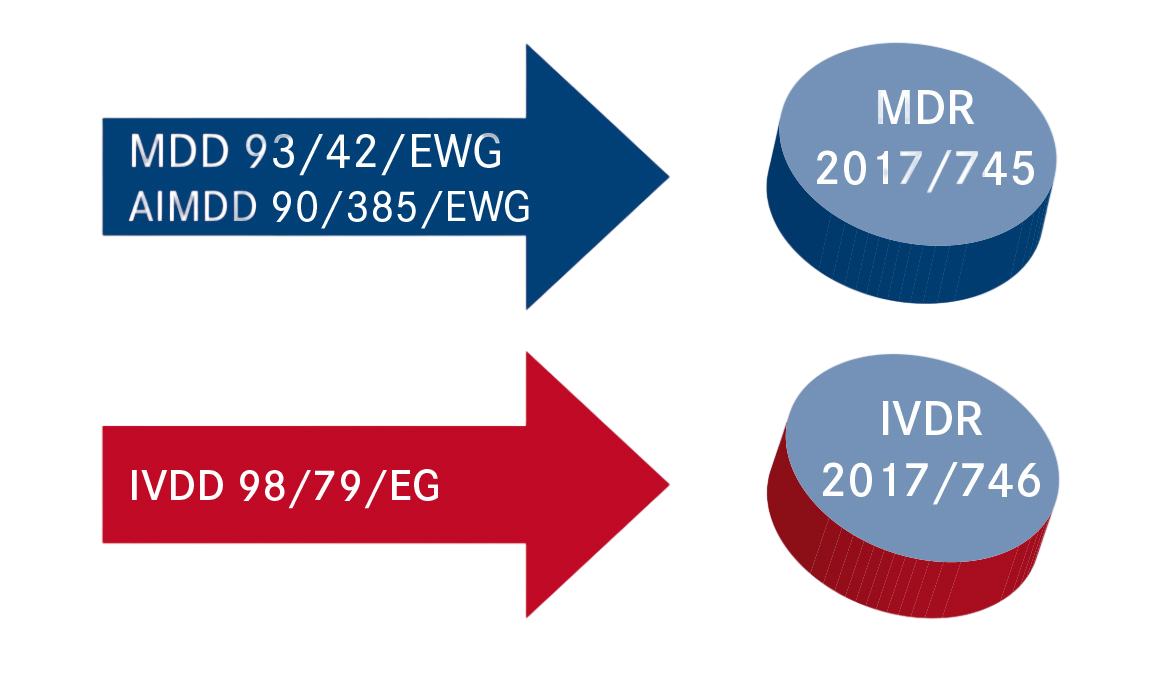
Bis zum 26. Mai 2021 galten in der EU zwei verschiedene Richtlinien für Medizinprodukte:
- Die Richtlinie 93/42/EWG über Medizinprodukte (MDD)
- Die Richtlinie 90/385/EWG über aktive Implantierbare medizinische Geräte (AIMDD)
Um den Rechtsrahmen für Medizinprodukte zu vereinheitlichen und zu vereinfachen, wurden die MDD und AIMDD durch eine einzige Verordnung ersetzt: die EU-Verordnung über Medizinprodukte (Verordnung EU 2017/745 MDR). Diese Verordnung betrifft alle Medizinprodukte außer In-vitro-Diagnostika, die durch eine separate Verordnung geregelt werden: die EU-Verordnung über In-vitro-Diagnostika (Verordnung EU 2017/746 IVDR). Die EU MDR ist am 25. Mai 2017 in Kraft getreten und seit dem 26. Mai 2021 anzuwenden während die IVDR seit dem 26. Mai 2022 anzuwenden ist.
Die MDR legt Regeln für das Inverkehrbringen, die Marktverfügbarkeit und die Inbetriebnahme von Medizinprodukten und deren Zubehör fest, die für den menschlichen Gebrauch bestimmt sind.
Diese Verordnung gilt auch für klinische Prüfungen, die innerhalb der EU durchgeführt werden und diese Medizinprodukte sowie das entsprechende Zubehör betreffen.
Die MDR ist umfassender und detaillierter als die MDD und die AIMDD. Dies bringt einige wichtige Änderungen für Hersteller, Benannte Stellen, Behörden und andere Akteure im Bereich der Medizinproduktebranche mit sich. Dazu gehören unter anderem:
- Eine erweiterte Definition von Medizinprodukten, die auch Produkte ohne medizinische Zweckbestimmung umfasst
- Ein strengeres Konformitätsbewertungsverfahren mit stärkerer Beteiligung von Benannten Stellen
- Ein System zur Identifizierung und Rückverfolgung von Produkten (UDI – Unique Device Identifier)
- Die Eingabe umfangreicher Daten in die EUDAMED-Datenbank
- Anforderungen an den Vertrieb von Medizinprodukten über das Internet bzw. an deren Fernabsatz
- Erhöhte Anforderungen an die klinische Bewertung und Nachbeobachtung nach dem Inverkehrbringen
- Erweiterte Haftungsregeln für Importeure und Händler
Unterschied zwischen den europäischen Richtlinien (MDD/AIMDD) und der Verordnung (MDR)
Der wesentliche Unterschied zwischen den europäischen Richtlinien (MDD/AIMDD) und der Verordnung (MDR) besteht darin, dass Richtlinien von den Mitgliedstaaten in nationales Recht umgesetzt werden müssen, während Verordnungen direkt und einheitlich in allen Mitgliedstaaten gelten und nationale Gesetze in den Bereichen ersetzen oder außer Kraft setzen, in denen diese Bestimmungen enthalten, die nationale Gesetze ersetzen oder überschreiben. In Deutschland wird die Umsetzung der MDR durch nationale Gesetze geregelt. Darüber hinaus wird die MDR auch durch das Medizinprodukterecht-Durchführungsgesetz (MPDG) ergänzt.
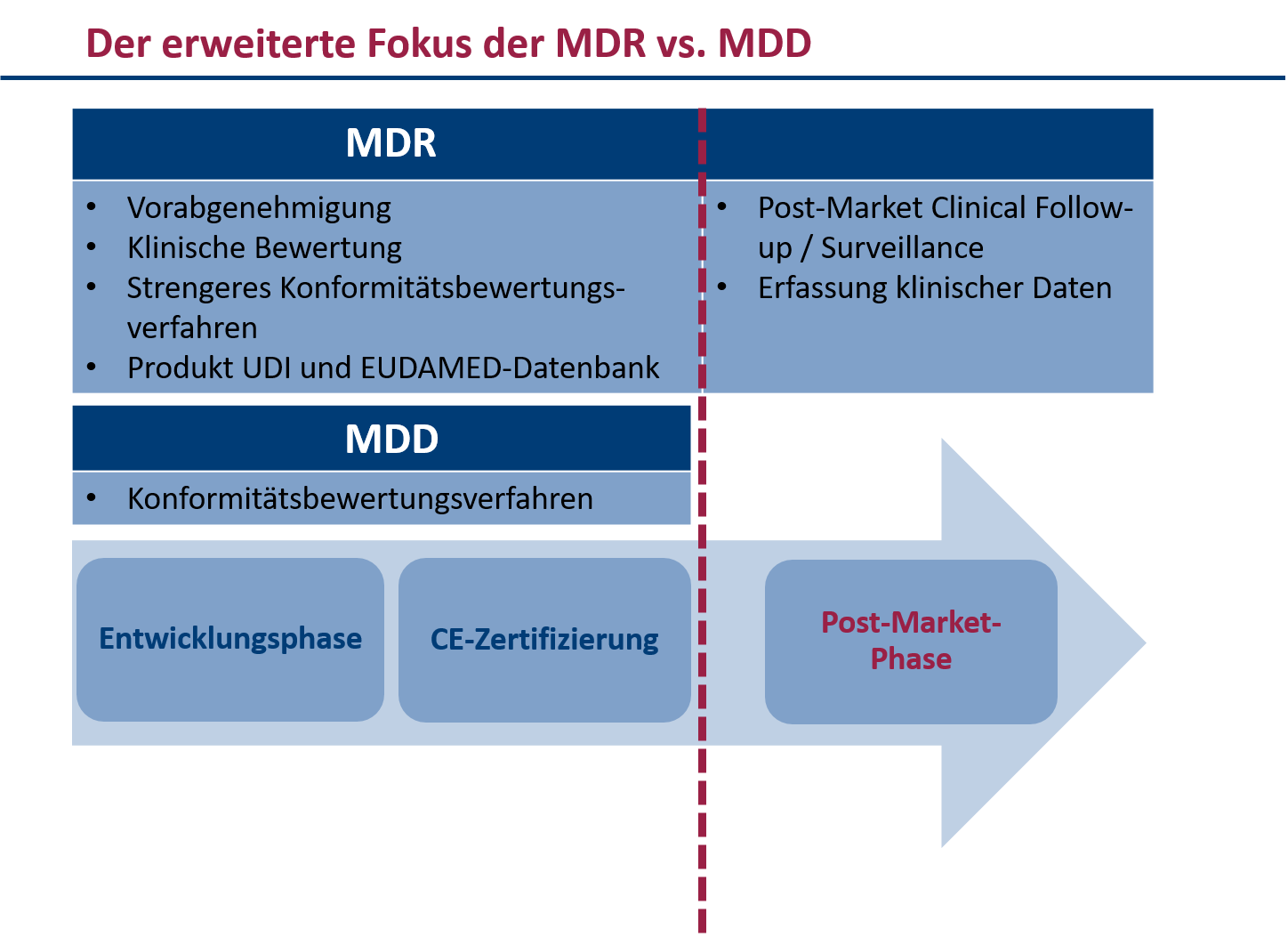
Sammlung klinischer Daten nach dem Inverkehrbringen (Post-Market Clinical Follow-up)
Die MDR verstärkt die Anforderungen an die Post-Market Surveillance (PMS) und das Post-Market Clinical Follow-up (PMCF) von Medizinprodukten. Die Hersteller müssen für jedes Medizinprodukt ein PMS-System einrichten, um die Sicherheit und Leistung des Produkts während seines gesamten Lebenszyklus zu bestätigen. Außerdem müssen Hersteller zukünftig mehr klinische Daten sammeln und auswerten, um Nebenwirkungen und Kontraindikationen zu ermitteln und ein angemessenes Nutzen-Risiko-Verhältnis des Produkts zu gewährleisten. Die Datenqualität und -verwaltung muss ebenfalls optimiert werden.
Übergangsfristen von den europäischen Richtlinien (MDD/AIMDD) zur Verordnung (MDR)
Neue Produkte unterliegen ab Beginn des Zertifizierungsprozesses dem vollen Geltungsbereich der MDR. Für bereits existierende Produkte bestehen unterschiedliche Übergangsfristen. Diese können u. a. von der Einstufung der Risikoklasse nach MDD/MDR abhängen. Nach dem von der Europäischen Kommission am 6. Januar 2023 angenommenen Vorschlag zur Änderung der MDR und IVDR gelten nun folgende Übergangsfristen:
- Die Übergangsfristen für Medizinprodukte, die vor dem 26. Mai 2021 ein Zertifikat oder eine Konformitätserklärung erhalten haben, werden um ein Jahr verlängert. Das bedeutet, dass die Übergangsfrist nun bis zum 26. Mai 2025 oder bis zum Ablauf des Zertifikats/der Konformitätserklärung gilt.
- Die Übergangsfristen für benutzerdefinierte implantierbare Produkte der Klasse III und Produkte mit mittlerem und geringerem Risiko bleiben unverändert bei zum 26. Mai 2026 bzw. 31. Dezember 2028.
- Die Abverkaufsfristen für Medizinprodukte mit einem Zertifikat oder einer Konformitätserklärung vor dem 26. Mai 2021 werden abgeschafft, d. h. diese Produkte können verkauft werden, solange sie auf Lager sind.
Diese Änderungen sollen den Herstellern mehr Zeit und Flexibilität geben, um sich an die neuen Anforderungen der MDR anzupassen und die Versorgungssicherheit zu gewährleisten. Der Vorschlag wurde vom Europäischen Parlament und dem Europäischen Rat am 16. Februar 2023 angenommen.
Im folgenden Entscheidungsdiagramm können Sie nachvollziehen, welche Übergangsfristen für Ihr Medizinprodukt gelten.
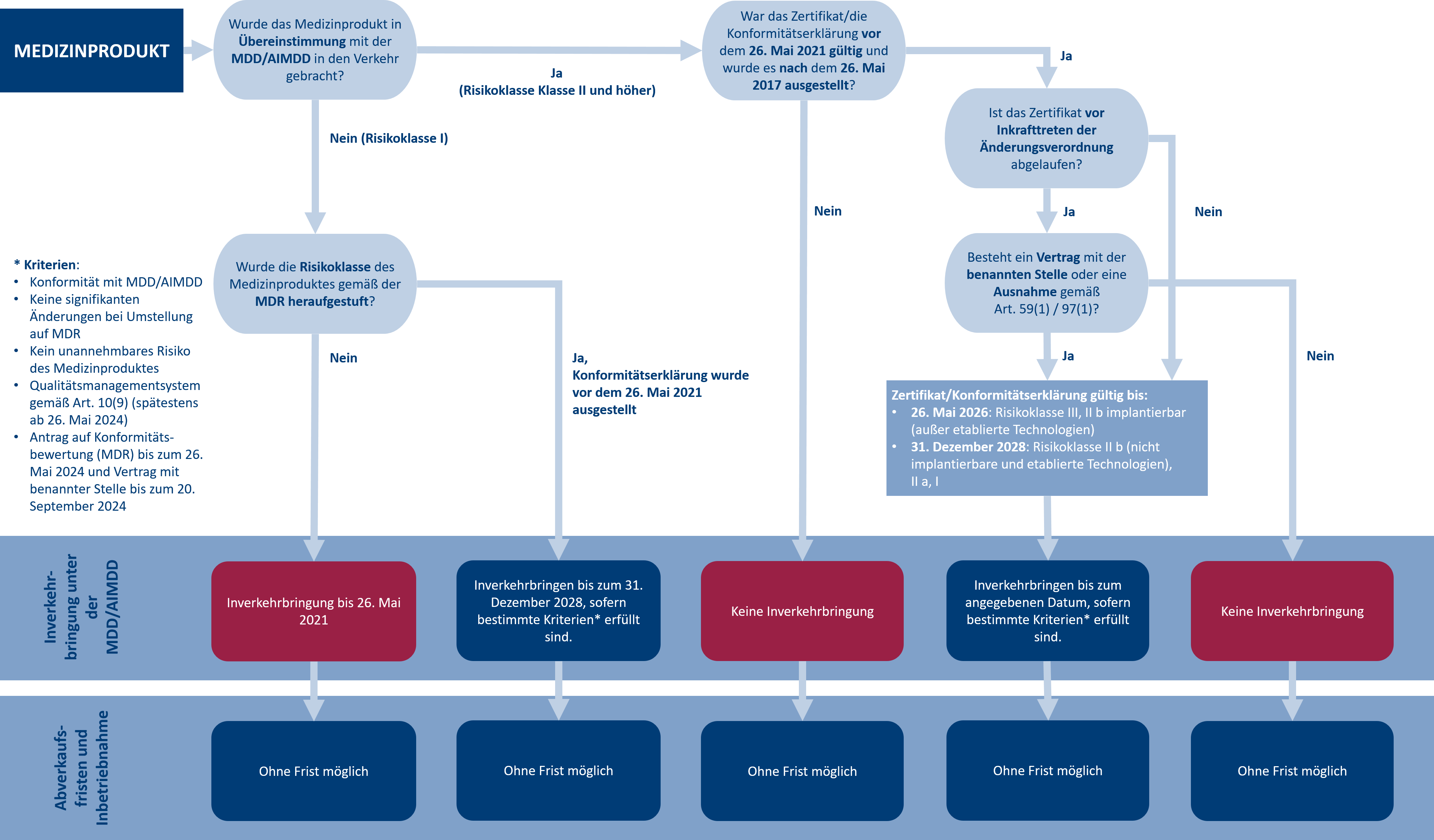
Die MDR verpflichtet außerdem alle Hersteller, eine Post-Market Surveillance (PMS) und das Post-Market Clinical Follow-up (PMCF) ihrer Medizinprodukte einzuführen. Die Prozesse sind für alle Medizinprodukte ab dem Inkrafttreten der MDR verpflichtend und haben keine Übergangsfristen.
Die Nutzung der EUDAMED-Datenbank bleibt hingegen freiwillig und hat daher auch keine Übergangsfrist.
Weitere relevante Dokumente für die Bestimmung der Übergangsfristen von Medizinprodukten finden Sie hier:
- FAQ zu den Übergangsfristen, welches von dem Arbeitskreis NAKI (Nationaler Arbeitskreis zur Implementierung von MDR und IVDR) bereitgestellt wird und von dem Bundesministerium für Gesundheit (BMG) ins Leben gerufen wurde
- FAQ von der CAMD Transition Sub Group (CAMD: „Competent Authorities for Medical Devices“)
- Dokument 2019-10 von der MDCG (Medical Device Coordination Group)
In diesem Dokument werden Überlegungen zum Übergang bezüglich der Marktüberwachung dargelegt. Demnach ist es möglich, dass ein Hersteller von den Übergangsfristen auch dann profitiert, wenn seine Benannte Stelle noch nicht für die MDR benannt ist. Die Voraussetzung ist aber, dass die Benannte Stelle weiterhin die Marktüberwachung aktiv durchführt und bei Problemen handelt.
Anforderungen an Medizinprodukte nach EU-MDR
Die MDR enthält viele neue Vorgaben an die Hersteller von Medizinprodukten, um die Qualität und Sicherheit der Produkte für die Nutzer zu gewährleisten. Zu den wichtigsten gehören die folgenden Punkte:
- Mehr und bessere Sicherheits– und Leistungsanforderungen sowie einen höheren Umfang der Begleitinformationen für Patient:innen
- Eine detaillierte und aktuelle Technische Dokumentation
- Regelmäßige Überwachung des Medizinproduktes nach dem Inverkehrbringen durch die Post-Market-Surveillance
- Eine notwendige klinische Bewertung und –Prüfung mit klaren Vorgaben an die erhoben klinischen Daten
- Besondere Anforderungen an Medizinprodukte mit Gefahrstoffen
- Eindeutige Identifizierung jedes Produkts mittels einer Produktidentifikationsnummer (UDI)
Technische Dokumentation
Ein wichtiger Teil der einzureichenden Unterlagen ist die technische Dokumentation des Medizinproduktes. Diese muss eindeutig, verständlich und gut gegliedert sein. Die technische Dokumentation sollte grundlegenden Sicherheits- und Leistungsanforderungen enthalten und ist von zentraler Bedeutung für die Zertifizierung eines Medizinproduktes.
Die hier aufgeführten Elemente müssen enthalten sein. Weitere Einzelheiten finden Sie in Anhang II.
Klassifizierung, Zulassung, Inverkehrbringen
Zusätzlich zu den Anforderungen an die technische Dokumentation hat die neue EU-MDR auch Auswirkungen auf die Klassifizierung, Zulassung und Vermarktung eines Medizinprodukts:
- Durch die MDR ändern sich die Risikoklassen einiger Medizinprodukte. Die Klassifizierung von alten und neuen Produkten muss den neuen Richtlinien angepasst werden.
- Das Konformitätsbewertungsverfahren aus der MDD gibt es in der neuen MDR nicht mehr und wird durch eine Konformitätsbewertung durch den Hersteller, also eine Zulassung ohne eine europäische Behörde für Medizinprodukte, abgelöst.
- Die MDR führt zu einer EU-weiten Vereinheitlichung der Tätigkeit und der Prüfbescheinigungen der Benannten Stelle (MDR Zertifikat). Zusätzlich wird für die Benannten Stellen ein Scrutiny-Verfahrenen eingeführt. Benannte Stellen können somit verpflichtet werden, jeden neuen Antrag auf Konformitätsbewertung für ein Produkt mit hohem Risiko an eine Expertenkommission (die Medical Device Coordination Group, MDCG) zu melden.
Organisatorische Anforderungen in der MDR
Neben diversen Anforderungen an das Medizinprodukt selbst, bringt die EU-MDR außerdem auch einige neue Bestimmungen und Vorgaben für den Hersteller und sein Unternehmen mit sich. Zu den wichtigsten organisatorischen Neuerungen gehören:
- Die Aufbewahrungsdauer der Dokumentation steigt von 5 auf 10 Jahre.
- Qualitätsmanagementsystem (QM-System): Die Anforderungen der ISO 13485:2016- müssen erfüllt werden,unabhängig von der Risikoklasse eines hergestellten Medizinproduktes.
- Die MDR (und IVDR) führen erstmalig eine verantwortliche Person „für die Einhaltung der Regulierungsvorschriften“ ein, welche die Aufgaben des Sicherheitsbeauftragten übernimmt. Sowohl die Hersteller als auch deren Bevollmächtigte müssen eine solche Person Responsible for Regulatory Compliance (PRRC) benennen.
- Die Datenbank EUDAMED wird vergrößert und gewährt zusätzlich zu staatlichen Institutionen, auch Herstellern, Benannten Stellen sowie der Öffentlichkeit Zugriff.
- Die MDR unterscheidet zwischen mehreren Wirtschaftsakteuren, mit spezifischen Anforderungen an Hersteller, Händler, Importeure und Bevollmächtigte.
Aufbau der MDR
Im Vergleich zur MDD wurde die MDR formal und inhaltlich neu strukturiert. Im Folgenden haben wir für Sie Kapitel und Anhänge der MDR aufgelistet und alle relevanten Links zu den Originaltexten der MDR zusammengestellt.
MDR-Kapitel
- Kapitel: Anwendungsbereich und Definitionen
- Kapitel: Anforderungen an Hersteller, Distributoren und Mitgliedsstaaten: Konformitätsbewertungsverfahren, Labeling, Post-Market Clinical Follow-up, Post-Market Surveillance u. v. m.
- Kapitel: Nachverfolgbarkeit von Produkten, v. a. UDI
- Kapitel: Anforderungen an die benannten Stellen
- Kapitel: Klassifizierung und Konformitätsbewertung
- Kapitel: Klinische Bewertungen und klinische Prüfungen
- Kapitel: Marktüberwachung, Meldewesen
- Kapitel: Zusammenarbeit von Mitgliedsstaaten, „Medical Device Coordination Group“ und anderen Experten
- Kapitel: Vertraulichkeit, Datenschutz, Strafen
- Kapitel: Übergangsfristen und mehr
MDR-Anhang
- Allgemeine Sicherheits- und Leistungsanforderungen
- Technische Dokumentation
- Technische Dokumentation zur Überwachung nach dem Inverkehrbringen
- EU-Konformitätserklärung
- CE-Kennzeichnung für die Konformität
- Informationen, die bei der Registrierung von Produkten und Wirtschaftsakteuren vorzulegen sind (UDI)
- Anforderungen an die benannten Stellen
- Kriterien für die Klassifizierung
- Konformitätsbewertung auf der Grundlage eines Qualitätsmanagementsystems und Bewertung der technischen Dokumentation
- Konformitätsbewertung auf der Grundlage einer Baumusterprüfung
- Konformitätsbewertung auf der Grundlage einer Überprüfung der Produktkonformität
- Verfahren für Sonderanfertigungen
- Von einer benannten Stelle ausgestellte Bescheinigungen
- Klinische Bewertung und PMCF
- Klinische Untersuchungen
- Liste der Produktgruppen ohne medizinische Zweckbestimmung
- Entsprechungstabelle
Download-Bereich
| Originalversion der EU |
| Deutsch (HTML) |
| Englisch (HTML) |
| DEUTSCH (PDF) |
| ENGLISCH (PDF) |
Ihre Checkliste für den Umstieg von der MDD auf die MDR
✔ Befassen Sie sich mit den Inhalten der MDR (Siehe DOWNLOAD-BEREICH) und machen Sie sich mit den zukünftig verpflichtenden Verfahren vertraut.
✔ Prüfen Sie Ihr gesamtes Produkt-Portfolio auf den Aktualisierungsbedarf im Hinblick auf die neuen Anforderungen.
✔ Aktualisieren Sie die Technische Dokumentation entsprechend der Anforderungen der MDR.
✔ Prüfen Sie, welche klinischen Daten zum Nachweis der Anforderungen der MDR für die einzelnen Produktgruppen fehlen. Diese können ggf. noch im Rahmen von PMCF-Studien nach alter Richtlinie erhoben werden.
✔ Prüfen Sie Ihre Qualitätsmanagement-Dokumentation kritisch im Hinblick auf die neuen MDR-Anforderungen. Überlegen Sie, wie die Rückverfolgbarkeit in der gesamten Herstellungs- und Lieferkette sichergestellt werden kann.
✔ Erstellen Sie einen Plan, mit dem Sie die PMS der Produkte gewährleisten können. Ein zentraler Punkt der MDR ist die Überwachung der Produkte nach der Marktzulassung. Daraus leiten sich auch Folgen für die Produkthaftung ab.
✔ Da die MDR eine eindeutige Produktidentifikation erfordert, sollten Sie sich frühzeitig Gedanken über dessen Anbringung und Zuweisung machen.
✔ Die Aufgaben des bisherigen Sicherheitsbeauftragten werden durch die MDR erheblich erweitert. Die Verordnung verweist auf eine für die Einhaltung der Vorschriften verantwortliche Person mit sehr weitreichender Verantwortung für die Einhaltung der Vorschriften bei Herstellern und Bevollmächtigten.
✔ Berücksichtigen Sie bei Ihrer Zeit- und Budgetplanung die notwendigen personellen Ressourcen und Qualifikationsanforderungen für Ausbildung und einschlägige Berufserfahrung.
✔ Weitere Informationen zur MDR finden Sie in den Leitfäden der Medical Device Coordination Group (MDCG guidance documents), die zu verschiedenen Themenbereichen veröffentlicht wurden. Diese Leitfäden sind nicht rechtsverbindlich, sondern geben Empfehlungen auf EU-Ebene wieder.
Es besteht sofortiger Handlungsbedarf!
Benötigen Sie hierbei Unterstützung oder haben Sie Interesse an individueller Beratung, damit Sie die MDR spezifisch für Ihre Produkte erfolgreich umsetzen können?
Kontaktieren Sie uns noch heute unter info@mic-mainz.de oder +49 6131 17-9646 und lassen Sie sich umfassend von unseren Expert:innen beraten.